Introduction
The ongoing highly pathogenic avian influenza (HPAI) H5N1 pandemic, caused by the sublineage of clade 2.3.4.4b, has been spreading rapidly worldwide since 2021. Unlike other legacy H5 clades, this particular clade of H5 viruses has evolved through exceptional reassortment compatibility, resulting in the generation of many genotypes in nature. This virus infects wild birds, poultry, and mammals, including cats and dairy cattle (Plaza et al., 2004). A few human infections have recently been reported to be associated with exposure to poultry or dairy cattle (CDC, 2024). The detection of highly pathogenic avian influenza A virus (HPAIV) H5N1, especially in mammalian species, has raised concerns about its pandemic potential. No cases of human-to-human transmission of HPAI H5N1 have been reported to date, but we cannot rule out the possibility of its evolution through dynamic exchanges of gene segments between host species and different regions, considering the frequent reassortment events of the virus.
The clade 2.3.4.4b H5N1 viruses have been continuously introduced and circulated in Korea. According to numerous reports, these viruses were introduced to South Korea through the migration of wild birds, reassorted with Eurasian low-pathogenicity avian influenza (LPAI) viruses (Lee et al., 2023; Seo et al., 2024). Respiratory infections caused by the HPAIV H5N1 were observed to cause feline mortality in an animal shelter in Seoul, South Korea (Lee et al., 2024). The HPAIV H5N1 isolated from cats in Korea was a reassortant between the LPAI-Eurasian lineages and the HPAI-origin Chinese genotype G10. Genetic analyses indicated that H5N1 viruses have predominantly undergone reassortment with Eurasian lineage wild-bird LPAI viruses. Therefore, monitoring the reassortment of HPAI viruses and their international spread is important to proactively reduce the potential risks to animal and public health.
Materials and Methods
Ethics statement
The samples used in this study were collected passively from rescued or deceased wild animals at the Jeju Wildlife Rescue Center. The H5N1 avian influenza A virus was confirmed by the National Institute of Wildlife Disease Control and Prevention, Republic of Korea (21RW-01330), through an official reporting system.
Sampling
A total of 2,371 wild bird and mammal swab samples were collected from rescued or deceased wild animals from January 2021 to December 2023 at the Jeju Wildlife Rescue Center in Jeju Island, South Korea. Oral and rectal/cloacal swabs were collected in transport medium (Universal Transport Medium, Noble Biosciences™, Hwaseong, Korea) from the animals.
RNA extraction and reverse transcription polymerase chain reaction
RNA was extracted from swab samples using TRIzol LS (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNA was synthesized with an M-MLV reverse transcriptase kit (Invitrogen) using random hexamers according to the manufacturer’s protocol. Influenza A virus was detected using polymerase chain reaction (PCR) specific for the matrix gene, as previously described by the World Health Organization Collaborating Center for Reference and Research on Influenza, National Institute of Infectious Diseases, Gakuen 4-7-1, Musashi-Murayama-shi, Tokyo, Japan.
Virus isolation
The raw sample that tested positive for influenza A virus-specific reverse transcription PCR (RT-PCR) was centrifuged at 3,000 × g at 4°C for 10 minutes. The clear supernatants were filtered through a 0.22 µm filter before inoculation into 10-day-old embryonated chicken eggs. Harvested allantoic fluid was tested for hemagglutinin (HA) activity using RT-PCR.
Genomic sequencing
The total RNAs extracted from the viral isolate were submitted to Macrogen, Inc. (Seoul, Korea) for transcriptome resequencing. A cDNA library was constructed using the TruSeq Stranded Total RNA Library Prep Gold Kit (Illumina Korea, Seoul, Korea), followed by high-throughput sequencing on an Illumina platform with 101 bp paired-end reads. Raw FASTQ files were imported into Geneious Prime (version 2024.0.3; Dotmatics, Boston, MA, USA) using paired-end reads. The reads were trimmed using the BBDuk plugin version 38.84 (DOE Joint Genome Institute, CA, USA) with a cutoff threshold for the average base quality score set at 30 (99.9% base-call accuracy). The trimmed reads were imported into the QIAGEN CLC Genomics Workbench (v21.0.5; Qiagen, Hilden, Germany) for genomic assembly. The resulting contig sequences with predicted influenza virus lengths were aligned to H5N1 reference sequences. The contigs were confirmed by mapping the trimmed reads. Consensus sequences were aligned with de novo contigs to generate newly assembled segments.
Sequence analysis and phylogeographic inference
We utilized the GISAID database ( https://gisaid.org; Elbe & Buckland-Merrett, 2017; Khare et al., 2021; Shu & McCauley, 2017) to identify the highest similarities for each segment of the H5N1 virus isolate in this study, A/Northern Shoveler/Jeju/D60/2023. The reference sequences for the phylogenetic analysis were collected from the 2.3.4.4b clade H5N1 viruses between 2022 and 2024, which were closely related to the isolate in this study.
The overall phylogeographic inference method used in this study was primarily based on the practical guide of Dellicour et al. (2021) and the Bayesian statistical framework implemented in BEAST v10.5.0-beta3 (Suchard et al., 2018). Multiple sequence alignments were performed using the MAFFT algorithm (Katoh & Standley, 2013) in Geneious Prime (v2023.2.1) from the collected datasets of HA and NA genes with location traits. The aligned sequences were further tested to exclude potential recombinants using the recombination detection program RDP4 (Martin et al., 2015). The datasets were further optimized to improve the temporal signal using TempEst v.1.7.2 analysis of the maximum likelihood tree generated with FastTree (Price et al., 2010; Rambaut et al., 2016).
The substitution models selected for the HA and NA sequences were TN93+F+G4 and HKY+F, respectively, using ModelFinder (Kalyaanamoorthy et al., 2017). Marginal likelihood estimation was performed using path sampling/stepping stone sampling (Baele et al., 2012; 2013), and the best log marginal likelihood score was obtained from 18 combinations of three site models for location traits, two clock models, and three tree prior models. The selected model combinations were the Cauchy RRW model for the location trait, uncorrelated relaxed clock model, and coalescent exponential model (Tree Prior) for HA sequences and the Cauchy RRW model for the location trait, strict clock model, and coalescent Bayesian Skygrid model (Tree Prior) for NA sequences.
Log and tree files were generated by running BEAST on a 500,000,000 Monte Carlo (MCMC) chain. Effective sample size, acceptable through Tracer v1.7.2 maximum clade credibility (MCC) trees, was obtained with TreeAnnotator v10.5.0 (Rambaut et al., 2018). The MCC trees were further visualized using SPREAD 4 for phylogeographic inference (Nahata et al., 2022).
The MCC trees for PB2, PB1, PA, NP, M, and NS were obtained using the Bayesian statistical framework implemented in BEAST. The substitution models for each dataset were selected and run with a 100,000,000 MCMC chain implemented using strict clock and coalescent constant models.
Results and Discussion
In this study, RT-PCR results indicate that only one sample tested positive for the influenza A virus. This sample was obtained from a Northern Shoveler that had died and was collected on Jeju Island, South Korea, on December 26, 2023. The Northern Shoveler, a migratory bird species in Korea, most likely contracted the influenza A virus from other migratory birds in a nearby flock. We isolated a HPAI H5N1 virus by inoculating it into embryonated chicken eggs. This isolate was designated A/Northern Shoveler/Jeju/D60/2023 (hereafter referred to as D60), and whole-genome sequencing was performed after the isolation. The hemagglutination assay titer of the isolate was 4,096. Nucleotide sequences were deposited in the GISAID database with the accession number EPI_ISL_19035743.
Six of the eight gene segments (PB2, PB1, PA, HA, NP, and MP) of D60 shared high nucleotide identities (ranging from 99.43% to 99.87%) with HPAI H5N1 viruses of clade 2.3.4.4b isolated from wild birds in Japan (Table 1). The HA gene segment has a multi-basic amino acid motif at the HA cleavage site, PLREKRRKR/G, which is similar to previously reported H5N1 viruses in Korea (Seo et al., 2024). The NA and NS gene segments were more closely related (99.77-99.93%) to those of isolates from British Columbia, Canada (Table 1), according to the GISAID BLAST results (as of April 1, 2024). Protein sequence BLAST results for the NA protein showed a 100% amino acid identity match with A/polar bear/Alaska/23-038123/2023 (accession number: EPI_ISL_18976667), an isolate from a deceased polar bear in Alaska, USA, in October 2023. Additionally, notable mutations were detected in the HA, NA, M, NP, NS, and PB2 gene segments of D60 (Table 2) using FluServer v1.0 in GISAID (Elbe & Buckland-Merrett, 2017; Khare et al., 2021; Shu & McCauley, 2017). Mutations associated with host specificity were observed in HA (N101S, with classical H3N2 numbering), M (S82N), and NP (N319K), whereas virulence-associated mutations were observed in the NS1 (V226T), NS2 (E67G), and PB2 (K42R) genes.
As the genomic sequences of D60 were not closely related to recent H5N1 viruses reported in mainland Korea between 2022 and 2023, we performed phylogeographic inference analysis based on the HA and NA gene segments. The time-scale MCC tree of the HA gene segment (Fig. 1A) and phylogeographic analysis (Fig. 2) revealed that D60 was more closely related to H5N1 viruses detected in Japan between 2022 and 2023 than recent Korean H5N1 viruses. The time that the most recent common ancestor of the D60 HA gene segment with the closest sequence occurred was estimated to be in August 2023. The NA gene segment was more closely related to H5N1 viruses reported in Alaska and Canada of North America between 2022 and 2023; however, it did not belong to the lineage of 2.3.4.4b clade H5N1 viruses recently detected in dairy cows in the USA (Fig. 1B; Hu et al., 2024; Wang et al., 2024). The time their most common ancestor occurred was estimated to be in February 2023. The phylogeographic analysis further demonstrated that the NA gene segment of D60 originated in Alaska and Canada in North America (Fig. 3).
Phylogenetic analysis based on the PB2, PB1, PA, NP, and M segments (Figs. 4Fig. 5Fig. 6Fig. 7-8) revealed that D60 shared a recent common ancestor with H5N1 viruses (clade 2.3.4.4b) in Japan and was distantly related to recent Korean H5N1 viruses. In the NS segments, D60 was closely related to H5N1 viruses detected in Alaska and Canada, similar to the NA segments (Fig. 9). Collectively, D60 may be a reassortment of gene segments originating from the intercontinentally spread 2.3.4.4b H5N1 viruses between North America and East Asia. Previously, the transmission of LPAI virus and HPAIV by wild birds between the Eurasian continent and the Americas through the Pacific and North Atlantic flyways (Alkie et al., 2022) has been documented (Parums, 2023; Wang et al., 2024). Additionally, H5N1 viral transmission, particularly through migratory birds, between Canada and the USA has resulted in infection cases in wild coastal birds and marine mammals (Puryear et al., 2023). The D60 isolate in this study suggests a dynamic transmission of the H5N1 clade 2.3.4.4b HPAIV in the North Pacific region, encompassing North America, Japan, and Korea.
In conclusion, our study presents a new case of the HPAI H5N1 clade 2.3.4.4b virus in a wild bird with a different ancestral origin in Jeju Island. The viral genome was phylogenetically distinct from previous Korean H5N1 isolates from mainland South Korea in the winter of 2022-2023 and was closely related to the viruses reported in North America and Japan. This finding highlights the transmission route of HPAIV H5N1 between North America, Japan, and South Korea. Considering the recent pandemic of HPAI H5N1 in North America, there should be a collaborative monitoring of HPAIV H5N1 transmission and evolution among the countries within the transmission route.
Acknowledgments
We extend our gratitude to the experts at the National Institute of Wildlife Disease Control and Prevention and the Ministry of Environment in the Republic of Korea for their invaluable assistance in confirming the presence of HPAIV in our samples and providing guidance for the deposition of the samples. Their expertise and collaboration were instrumental in the progress of this study.
We gratefully acknowledge all data contributors, that is, the authors and the laboratories responsible for obtaining the specimens, generating the genetic sequence and metadata, and sharing via the GISAID Initiative, on which this research is based.
This study was supported by the Bio & Medical Technology Development Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT of Korea (NRF-2021M3E5E3083402). This study was supported by the Basic Science Research Program through the NRF, funded by the Ministry of Education of Korea (2020R1A6A1A06046235).
Author Contributions
Conceptualization: J. H. Jang, H. K. Kim. Data curation: V. T. Lo, D. Y. Mun, M. C. Kang. Formal analysis: V. T. Lo, S. S. Jang, D. Y. Mun. Funding acquisition: H. K. Kim. Investigation: J. H. Jang. Methodology: V. T. Lo, S. S. Jang. Project administration: H. K. Kim. Resources: J. H. Jang, Y. M. Yun. Software: V. T. Lo, M. C. Kang. Supervision: H. K. Kim, Y. M. Yun. Validation: S. S. Jang. Visualization: V. T. Lo, D. Y. Mun. Writing – original draft: V. T. Lo, H. K. Kim. Writing – review & editing: Y. M. Youn, S. Youk.
References
Centers for Disease Control and Prevention (CDC) (2024, Retrieved June 1, 2024) Technical report: june 2024 highly pathogenic avian influenza A (H5N1) viruses from https://www.cdc.gov/bird-flu/php/technical-report/h5n1-06052024.html
Figures and Tables
Fig. 1
Phylogenetic relationship between A/Northern Shoveler/Jeju/D60/2023 and recent HPAIVs of H5N1 clade 2.3.4.4b. (A) Time-scale MCC tree based on HA gene segments. (B) Time-scale MCC tree based on NA gene segments. The isolate in this study, A/Northern Shoveler/Jeju/D60/2023, is indicated in a red taxon. H5N1 HPAIVs recently reported from wild birds and domestic cats in Korea are indicated in blue taxa. The x-axis defines the time scale in decimal years. All the reference viruses belonged to the 2.3.4.4b clade. MCC, maximum clade credibility; HA, hemagglutinin; HPAIV, highly pathogenic avian influenza A virus.
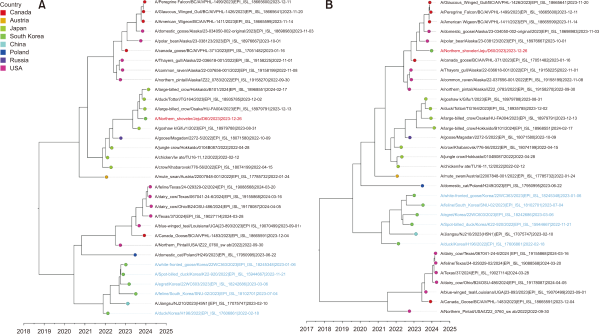
Fig. 2
Phylogeographic diffusion of recent HA gene segments of H5N1 2.3.4.4b clade. HA gene segment of A/Northen shoveler/Jeju/D60/2023 was inferred to have originated from Japan. All the reference viruses belonged to the 2.3.4.4b clade. HA, hemagglutinin. Source: Nahata et al. (2022).
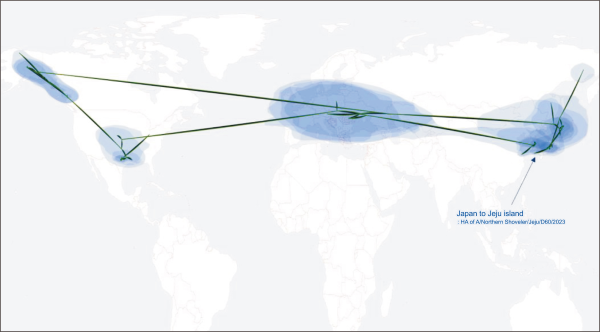
Fig. 3
Time-scale MCC tree based on PB2 gene segments. The time-scale MCC tree based on PB2 gene segments shows the phylogenetic relationships and temporal dynamics of the H5N1 strains in this study. A/Northern Shoveler/Jeju/D60/2023 is indicated by a red taxon, while the strains recently reported in South Korea are indicated by blue taxa. MCC, maximum clade credibility.
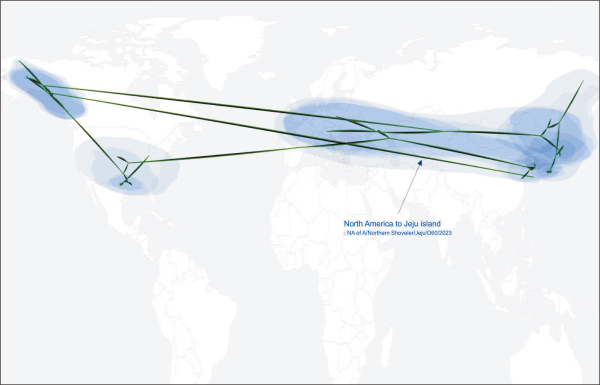
Fig. 4
Phylogeographic diffusion of recent NA gene segments of H5N1 2.3.4.4b clade. NA gene segment of A/Northen shoveler/Jeju/D60/2023 was inferred to have originated from North America. Source: Nahata et al. (2022).
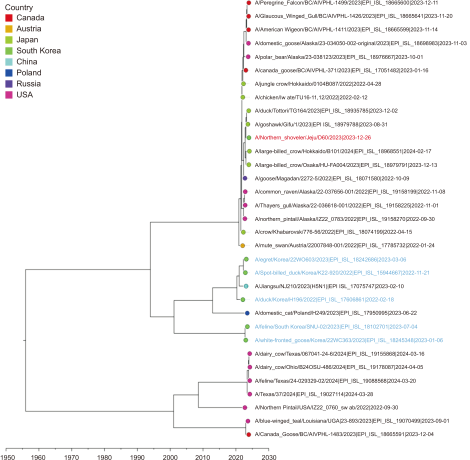
Fig. 5
Time-scale MCC tree based on PB1 gene segments. The time-scale MCC tree based on PB1 gene segments shows the phylogenetic relationships and temporal dynamics of the H5N1 strains in this study. A/Northern Shoveler/Jeju/D60/2023 is indicated by a red taxon, while the strains recently reported in South Korea are indicated by blue taxa. MCC, maximum clade credibility.
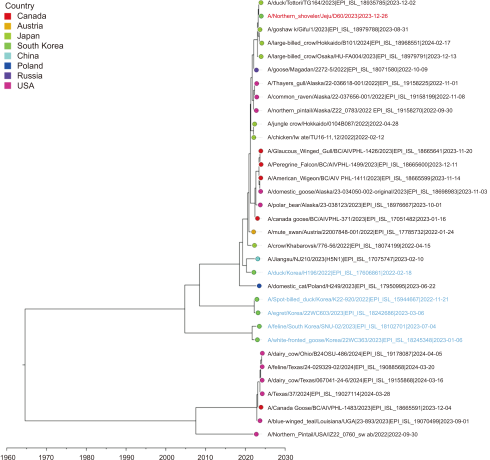
Fig. 6
Time-scale MCC tree based on PA gene segments. The time-scale MCC tree based on PA gene segments shows the phylogenetic relationships and temporal dynamics of the H5N1 strains in this study. A/Northern Shoveler/Jeju/D60/2023 is indicated by a red taxon, while the strains recently reported in South Korea are indicated by blue taxa. MCC, maximum clade credibility.
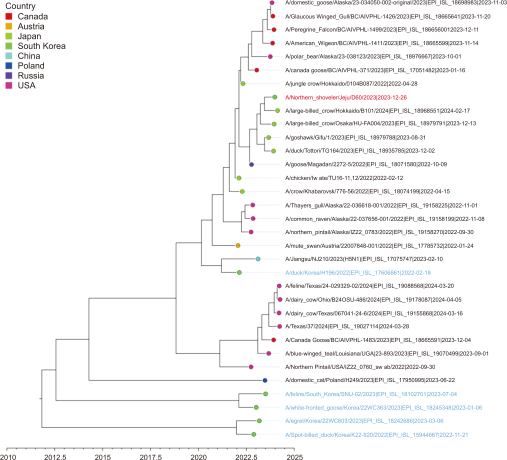
Fig. 7
Time-scale MCC tree based on NP gene segments. The time-scale MCC tree based on NP gene segments shows the phylogenetic relationships and temporal dynamics of the H5N1 strains in this study. A/Northern Shoveler/Jeju/D60/2023 is indicated by a red taxon, while the strains recently reported in South Korea are indicated by blue taxa. MCC, maximum clade credibility.
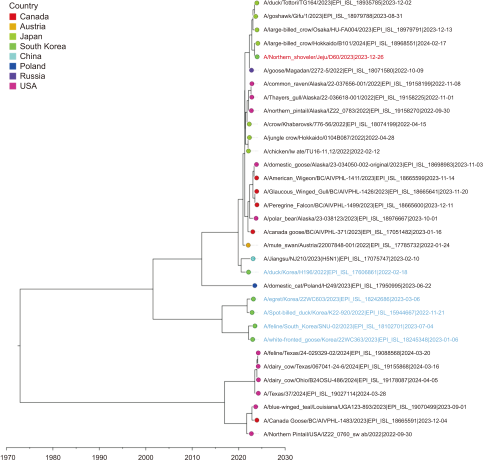
Fig. 8
Time-scale MCC tree based on M gene segments. The time-scale MCC tree based on M gene segments shows the phylogenetic relationships and temporal dynamics of the H5N1 strains in this study. A/Northern Shoveler/Jeju/D60/2023 is indicated by a red taxon, while the strains recently reported in South Korea are indicated by blue taxa. MCC, maximum clade credibility.
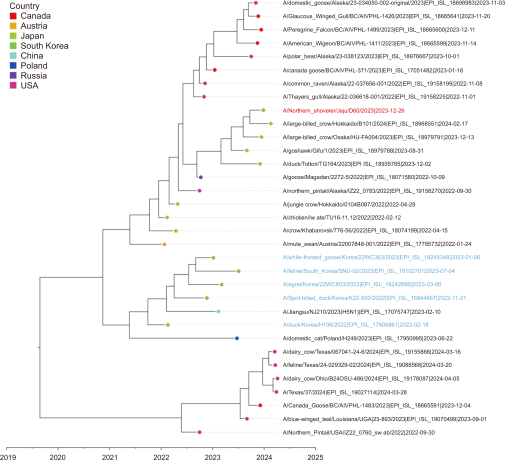
Fig. 9
Time-scale MCC tree based on NS gene segments. The time-scale MCC tree based on NS gene segments shows the phylogenetic relationships and temporal dynamics of the H5N1 strains in this study. A/Northern Shoveler/Jeju/D60/2023 is indicated by a red taxon, while the strains recently reported in South Korea are indicated by blue taxa. MCC, maximum clade credibility.

Table 1
Closest homologs to A/Northern Shoveler/Jeju/D60/2023 identified using GISAID (https://gisaid.org) BLAST
| Gene segment |
Strain with the highest identity | % Identity | Accession no. | Location |
|---|---|---|---|---|
| PB2 | A/duck/Tottori/NK40/2023 (A/H5N1) | 99.69 | EPI_ISL_18935784 | Japan/Tottori |
| A/large-billed crow/Hokkaido/20231207001/2023 | 99.69 | EPI_ISL_18740266 | Japan/Hokkaido | |
| PB1 | A/large-billed crow/Hokkaido/B114/2024 | 99.87 | EPI_ISL_19033210 | Japan/Hokkaido |
| A/goshawk/Gifu/1/2023 | 99.87 | EPI_ISL_18979788 | Japan/Mie | |
| PA | A/goshawk/Gifu/1/2023 | 99.73 | EPI_ISL_18979788 | Japan/Mie |
| A/duck/Tottori/TG164/2023 | 99.73 | EPI_ISL_18935785 | Japan/Tottori | |
| HA | A/duck/Tottori/TG164/2023 | 99.49 | EPI_ISL_18935785 | Japan/Tottori |
| A/blow fly/Kagoshima/23a738D/2023 | 99.43 | EPI_ISL_18969146 | Japan/Kagoshima | |
| NP | A/duck/Tottori/TG164/2023 | 99.68 | EPI_ISL_18935785 | Japan/Tottori |
| A/duck/Tottori/NK40/2023 (A/H5N1) | 99.68 | EPI_ISL_18935784 | Japan/Tottori | |
| NA | A/Glaucous_Winged_Gull/BC/AIVPHL-1426/2023 | 99.93 | EPI_ISL_18665641 | Canada/British Columbia |
| A/Mallard/BC/AIVPHL-1446/2023 | 99.93 | EPI_ISL_18665669 | Canada/British Columbia | |
| MP | A/large-billed crow/Osaka/HU-FA004/2023 | 99.80 | EPI_ISL_18979791 | Japan/Osaka |
| A/goose/Magadan/2272-5/2022 | 99.80 | EPI_ISL_18071580 | Russian Federation/Magadan Oblast | |
| NS | A/Red_Tailed_Hawk/BC/AIVPHL-892/2023 | 99.77 | EPI_ISL_18665576 | Canada/British Columbia |
| A/mute swan/Austria/22007848-001/2022 | 99.77 | EPI_ISL_17785732 | Austria/Bundesland Steiermark |
Table 2
Notable mutations (interest level ≥2) detected from FluServer v1.0 in GISAID (https://gisaid.org)
| Gene segments | HA | NA | M | NP | NS1 | NS2 | PB2 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutationsa | N110S (N101S)b |
T139P (T128P) |
N170D (N158D) |
T188I (T187I) |
I436V (I437V) |
S82N | N319K | V226T | E67G | K482R |
| Reported effect | Host specificity |
Antigenic drift/escape mutant | Drug resistance |
Host specificity | Virulence, host specificity | Virulence | Virulence | Virulence | ||
aNumbering by the amino acid sequence of each viral protein from A/northern shoveler/Jeju/D60/2023. bClassical H3N2 numbering-base. Source: Khare et al. (2021), Elbe and Buckland-Merrett (2017), Shu and McCauley (2017).